The ProteoCast Server allows you to rapidly and easily explore how and to what extent missense mutations affect protein function. ProteoCast relies on GEMME, a highly efficient, unsupervised, and fully interpretable variant effect predictor (Laine et al., 2019). By focusing on how protein residues segregate along the topology of evolutionary trees, GEMME has proven instrumental for studying protein stability, function, and disease mechanisms in several studies -- see, for instance, Tsuboyama et al., 2023. It was extensively tested against over 2.5M experimental measurements (Abakarova et al., 2023, Laine et al., 2019). It is among the top-performing methods on the widely adopted ProteinGym benchmark.
You want to know more, collaborate, or you encounter a problem with the server? Please feel free to contact us!
If you would like to provide feedback, you are welcome to do so via this google form.
Input
Sequence
Please provide a multiple sequence alignment in FASTA, A3M, or A2M format. The protein of interest should be the first one in the alignment and should not contain any gap. We recommend generating the alignment with the highly efficient MMseqs2-based protocol implemented in ColabFold for an optimal balance between speed and accuracy. Using this protocol, the input alignment should contain at least a couple hundred sequences to obtain reliable predictions (Abakarova et al., 2023).
Alternatively, you may provide a UniProt ID instead of an alignment file. In this case, the server will automatically retrieve the corresponding multiple sequence alignment in A3M format from the AlphaFold Database, which will be used as input for the prediction.
Mutations
Optionally, users may provide a list of known mutations to be mapped onto the predicted results. This input is used to generate an additional SNP heatmap, where raw model scores are displayed in gray tones and known variants are highlighted for interpretation.
The file must be provided in CSV format and contain two columns, separated by commas:
-
Mutation: Amino acid substitution using standard notation (e.g.,
A23T). - Phenotype: Experimental or annotated effect associated with the mutation.
File format example
Mutation,Phenotype
A23T,Increased Stability
G45D,Decreased Activity
Structure
Optionally, you can opt for mapping the results onto a 3D structure, and compute structure-informed predictions. The latter correspond to raw prediction scores weighted by relative solvent accessibility (RSA). The relevance of RSA in variant effect prediction has been shown in Tsishyn et al.
The simplest way to use this functionality is described below:
- The query sequence corresponds to a UniProt entry. You can indicate the UniProt identifier, and when available, ProteoCast will automatically retrieve the corresponding 3D model from the AlphaFold Database.
- The query sequence does not match a UniProt entry. You may generate a 3D model along with the alignment using ColabFold and upload it directly. You may also provide an experimental structure from the Protein Data Bank, or a custom 3D model. Any user-defined 3D structure or model should be in PDB format. You can also provide a complex or multimer; in that case, please indicate the chain of interest. Otherwise, chain A will be used by default.

When the query sequence from the MSA does not exactly match the PDB sequence, a pairwise sequence alignment is performed. Residues that remain unaligned are shown in gray on the structure, and their scores are not RSA-weighted.
Output
Please check our example result page.
Mutational landscape
The predicted mutational landscape is displayed as two interactive heatmaps, where each square corresponds to a given amino acid substitution at a given position. The heatmap dimension is thus 20 by the length of the query protein sequence. The heatmap RAW SCORES contains the numerical estimates predicted by GEMME. The darker the color, the more negative the score, and thus the stronger the predicted effect. The heatmap VARIANT CLASSES indicates whether each mutation is considered as neutral (blue), mild (pink) or impactful (red) by ProteoCast. If a 3D structure is provided, the user has the option to visualize STRUCTURE-AWARE SCORES, which represent raw scores weighted by the relative solvent accessibility of each residue. For pre-computed results on the Drosophila melanogaster, we optionally provide a third heatmap SNPs with the raw scores in grey tones and known SNPs highlighted in colors, blue for population polymorphisms and red for lethal mutations.

By hovering the mouse cursor on the representation, a contextual window will specify the mutation and give some information about it (predicted raw score, class...). You may zoom in a particular region of interest.
The horizontal bar at the bottom of the heatmap reflects the confidence in the predictions. Dark blue indicates reliable predictions, and white unreliable ones. We consider predictions as unreliable when evolutionary information derived from the input alignment is too scarce.
Input alignement quality assessment
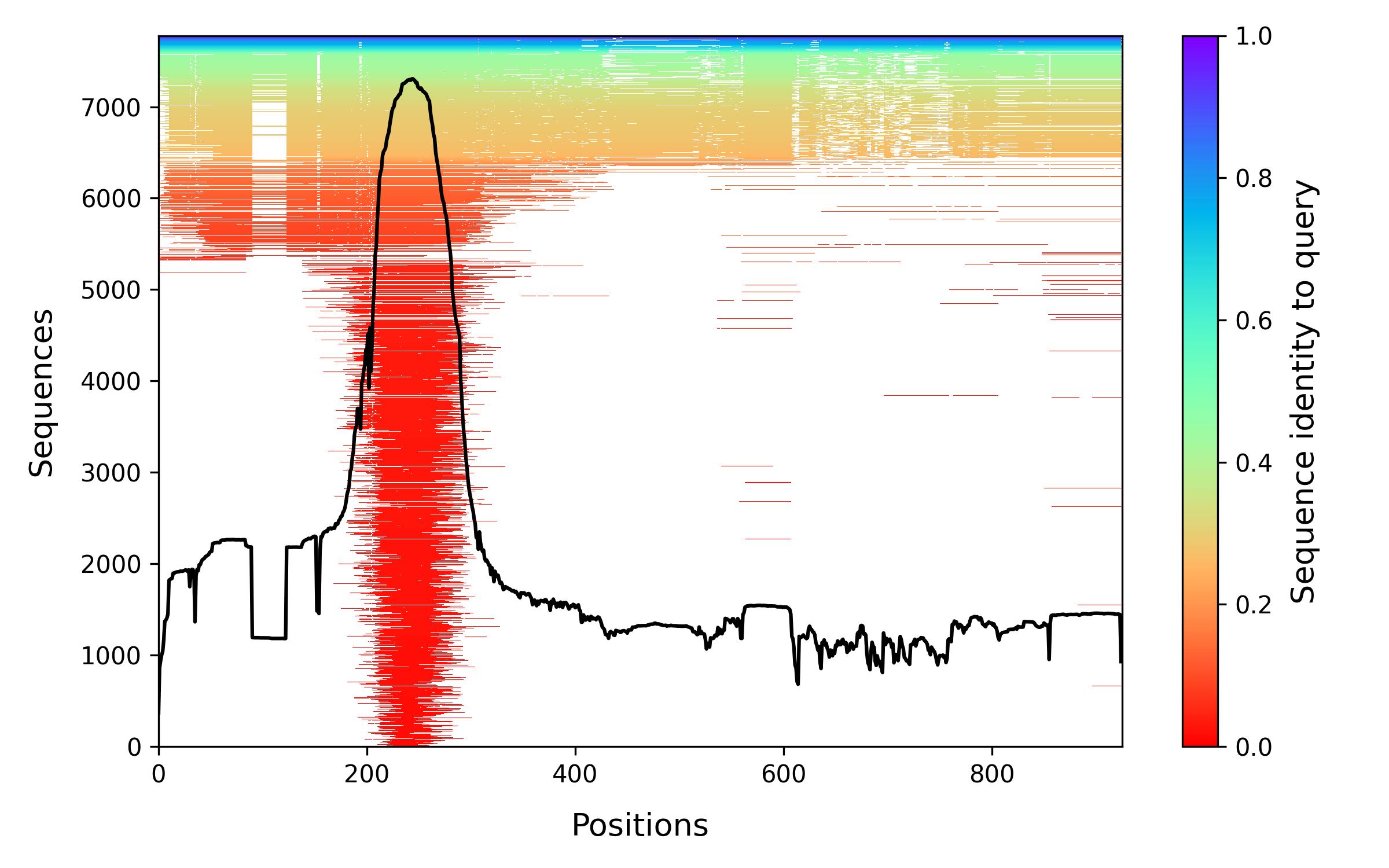
This plot gives an overview of the input alignment, where each horizontal line depicts a sequence and its color indicates its similarity with the query. Gaps correspond to white interruptions. The black curve reports the percentage of sequences that have an amino acid (as opposed to a gap) at each position of the query sequence. This representation is identical to the one used in ColabFold and we adapted the code from there.
Predicted score distributions for variant classification
This plot gives an overview of the predicted score distribution, fitted with a mixture of three Gaussians. To minimize biases, low-confidence predictions are excluded. At one end of the spectrum, mutations close to zero extending down to the median of the middle Gaussian are classified as neutral. At the other end, mutations with very negative scores are classified as impactful, until they are more likely to belong to the middle Gaussian than the leftmost one. Mutations falling in between are categorised as mild. The two vertical lines indicate the boundaries of the three classes.

Segmented mutational sensitivity profile
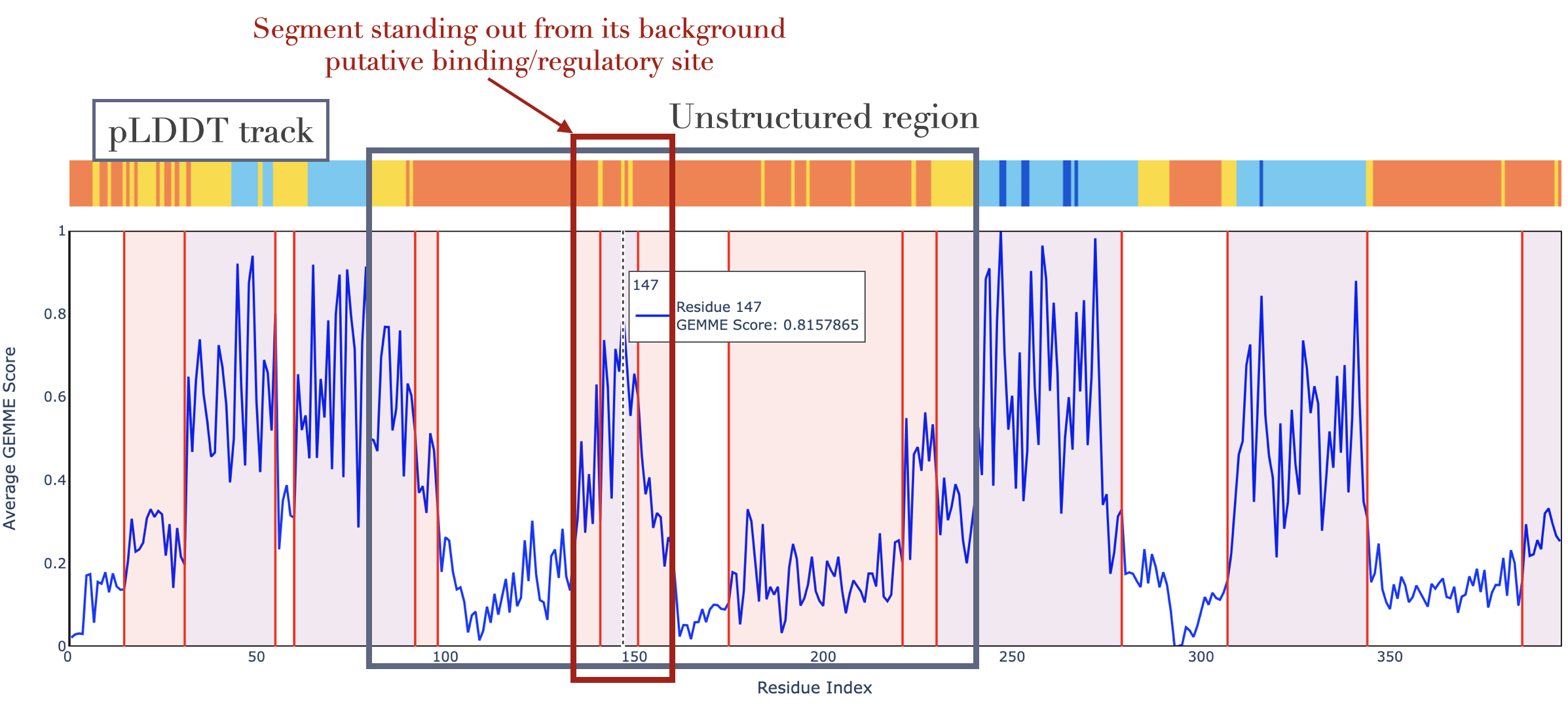
This interactive plot shows the per-residue mutational sensitivity, defined as the average GEMME score over the 19 possible substitutions. The values are scaled between 0 and 1. Residues highly sensitive to mutations will have a value close to 1, and residues highly tolerant a value close to 0. The profile is segmented using the FPOP algorithm, which identifies changepoints in the signal through 'functional pruning'. Informally, each detected changepoint signifies a shift in the mean. This representation allows for emphasizing protein segments under stronger or weaker selective pressure than their surrounding background. Purple: the segment mean is higher than the two neighbouring segments. Red: the segment mean is higher than one neighbour and lower than the other one. When an AlphaFold2-predicted structure is provided, a pLDDT track is added on top of the mutational sensitivity profile to improve interpretability -- for instance, toward the identification of putative binding and regulatory sites in unstructured regions. The four pLDDT classes are those defined in the AlphaFold Database, orange: very low, yellow: low, light blue: medium, dark blue: high. Note: a structure is considered as AlphaFold2-predicted if (i) retrieved from the AlphaFold Database, or (ii) TITLE ALPHAFOLD MONOMER V2.0 is present in the PDB file header, or (iii) alphafold / AF is present in the PDB file name (e.g., insulin_4459d_unrelaxed_alphafold2_ptm_model_1_seed_000.pdb or AF-P11362-F1-model_v4.pdb).
Mapping of predictions on the 3D structure
This interactive Molstar plugin allows you to explore the localisation of any mutation of interest, whether it is on the surface of the protein or within the core, whether it is part of a well-define secondary structure or an unstructured loop...etc. You can choose between different colour regimes that reflect the original B-factor values (pLDDT for AlphaFold models), the per-residue mutational sensitivity, or a binary classification of residues as tolerant or sensitive to mutations. Sensitive residues have more than half of the 19 possible substitutions being impactful. A fourth option, structure-aware sensitivity, integrates mutational scores with relative solvent accessibility for a more context-informed visualization. On each representation, you may visualise the segments located in unstructured regions (very low or low pLDDT) and whose sensitivity stands out against their surroundings. They are colored in purple or red, following the color scheme used for the segmented profile above. They are displayed using a cartoon-like representation in which the protein backbone is shown like a clay or putty structure. The size of the tube is proportional to the property being examined (pLDDT, mutational sensitivity or residue class). On the figure below, we can clearly see that the segment containing S146 has higher mutational sensitivity than its surroundings.

Downloaded output files
When downloading a completed ProteoCast job, all result files generated during the analysis are provided, including numerical predictions, intermediate data, and static visual summaries.
Main output files
4.QueryID_ProteoCast.csv-
Predicted effects for all possible single amino-acid substitutions.
Each row corresponds to one mutation and includes the following fields:
Field Description Mutation Amino-acid substitution in one-letter code (e.g. M1A).Variant_score Predicted impact score for the variant. More negative values indicate stronger deleterious effects. Residue Position of the mutated residue in the input sequence (1-based indexing). LocalConfidence Boolean flag indicating whether the residue is located in a region with sufficient evolutionary signal for reliable prediction. Variant_class Qualitative classification of the variant effect (e.g. neutral,mild,impactful).Residue_class Classification of the residue as sensitiveortolerant, based on the distribution of variant scores at this position.pdb_pos Corresponding residue index in the reference structure (empty if unmapped). Only included if structural information is used. RSA*Variant_score Structure-informed score obtained by weighting the Variant_score by the relative solvent accessibility (RSA) of the wild-type residue. Empty when structural information is unavailable. aligned Boolean flag indicating whether the residue is covered by the structure alignment. 8.QueryID_Segmentation.csv-
Segmentation of the protein sequence into contiguous regions based on mutational sensitivity profiles.
Each row corresponds to one segment and includes:
Field Description start Start position of the segment (inclusive). end End position of the segment (inclusive). mean Mean mutational sensitivity score over the segment. state Segment state (0, 1, 2), reflecting increasing levels of mutational sensitivity. type Metric used for segmentation ( GEMMEorpLDDT).protein Identifier of the analyzed protein. Segmentation is provided both for mutational sensitivity (
GEMME) and, when available, for AlphaFold confidence (pLDDT). rsa_values.csv- Computed relative solvent accessibility (RSA) values for all residues in the structure.
Complementary files
1.QueryID.fasta- Reference amino-acid sequence used for the analysis.
2.aliQueryID.fasta- Multiple Sequence Alignment (MSA) used for evolutionary inference, reformatted to FASTA when required.
mapping_fasta_pdb.csv- Mapping between sequence positions and structure residue indices.
14.QueryID_GEMME_pLDDT.csv- Combined GEMME evolutionary scores and AlphaFold pLDDT confidence values (when structural information is available).
7.QueryID_SNPs.csv(optional)-
These are the results of predictions for the mutations of interest provided by the user in the input file
mutantes.txt. Each mutation can optionally be associated with phenotypic annotations
Static figures
3.QueryID_msaRepresentation.jpg- Static visualization of MSA coverage and residue conservation along the sequence.
5.QueryID_MutLand.jpg- Static mutational landscape summarizing predicted effects across the protein.
6.QueryID_GMM.jpg- Gaussian Mixture Model visualization used to classify residues by mutational sensitivity.
9.QueryID_SegProfile.png- Static representation of the mutational sensitivity segmentation along the protein sequence.
Browser compatibility
The server's JavaScript code uses ECMAScript 2015 (ES6) features; therefore, it needs Chrome 58+, Edge 15+, Firefox 54+, Safari 10+, or Opera 55+. We have tested this server using the following browsers:
| OS | Version | Chrome | Firefox | Microsoft Edge | Safari |
| Linux | Ubuntu 22.04 | 131.0.6778.139 | 133.0.3 | n/a | n/a |
| MacOS | Mojave | 87.0.4280.88 | 83.0 | n/a | 14.0 |
| Windows | Windows 10 Home | 87.0.4280.88 | 84.0 | 87.0.664.60 | n/a |